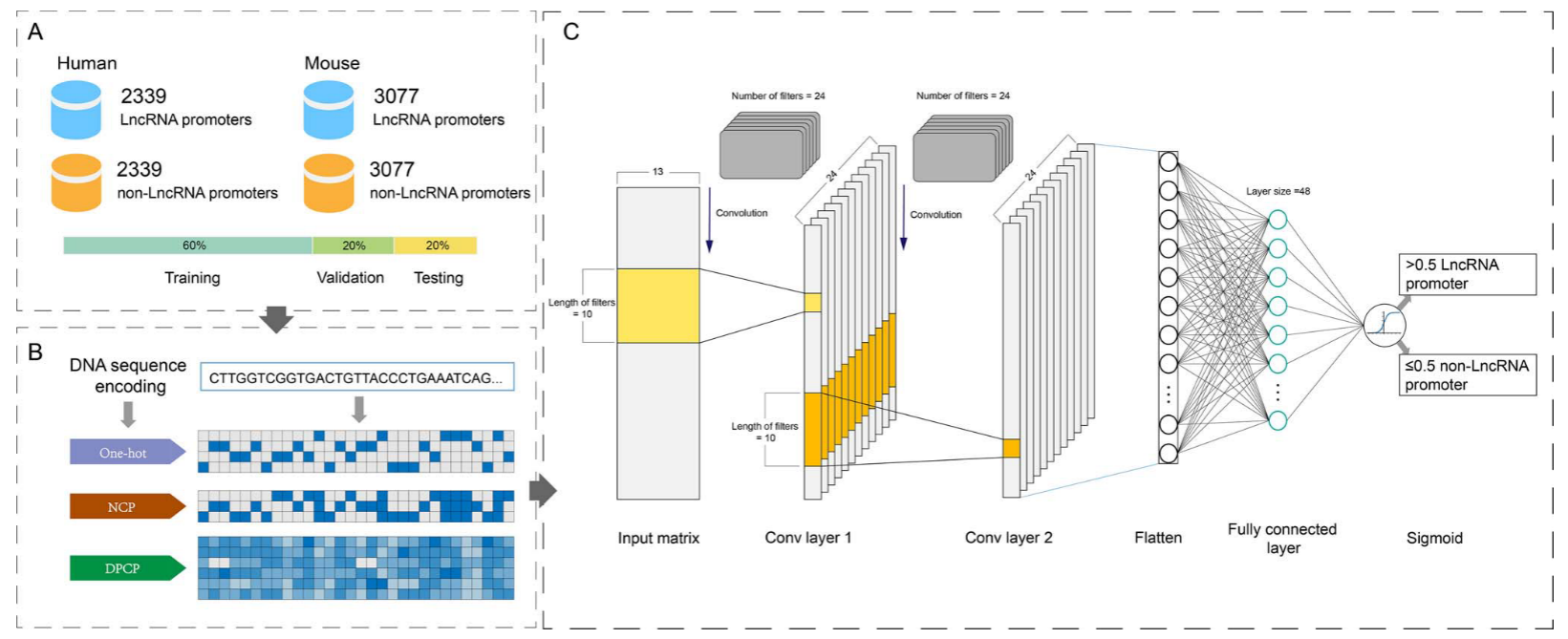
数据集是人和小鼠的lncRNA的启动子序列,获自真核启动子数据库(EPD)(考虑到RNA聚合酶通常结合在TSS的上游区域)

在TSS周围采集阳性样品,并含有更多的上游区域。从远离TSS的下游区域采集阴性样品。构建了7个基于序列长度从61到301 bp的数据集,步长为40 bp。对于具有nbp长度的序列的数据集,从TSS上游(n-20)bp至TSS下游20 bp提取阳性样品。以相同的方式提取阴性样品,阳性与阴性样品的比例保持在1:1。对于每个数据集,以6:2:2比例划分。
对数据的处理和特征提取,主要用了三种方式:1.one-hot 编码 2.NCP 3.DPCP。
1.独热编码方法可以有效地表示DNA序列并将脱氧核苷酸编码到二元载体中。在此基础上,将'A'编码为(1,0,0,0),'T'编码为(0,1,0,0),'G'编码为(0,0,1,0),'C'编码为(0,0,0,1)。因此,长度为L的DNA序列可以被转换成4×L矩阵A1。
2.四种脱氧核糖核苷酸携带不同的碱基,其在环结构、氢键强度和化学功能上不同。
3.连续脱氧核苷酸组合具有不同的物理化学性质,这是基因组功能元件鉴定的重要特征,并已用于启动子预测。在这项研究中,六个DPCP,即扭曲,倾斜,滚动,移位,滑动和上升,被用来编码DNA序列。
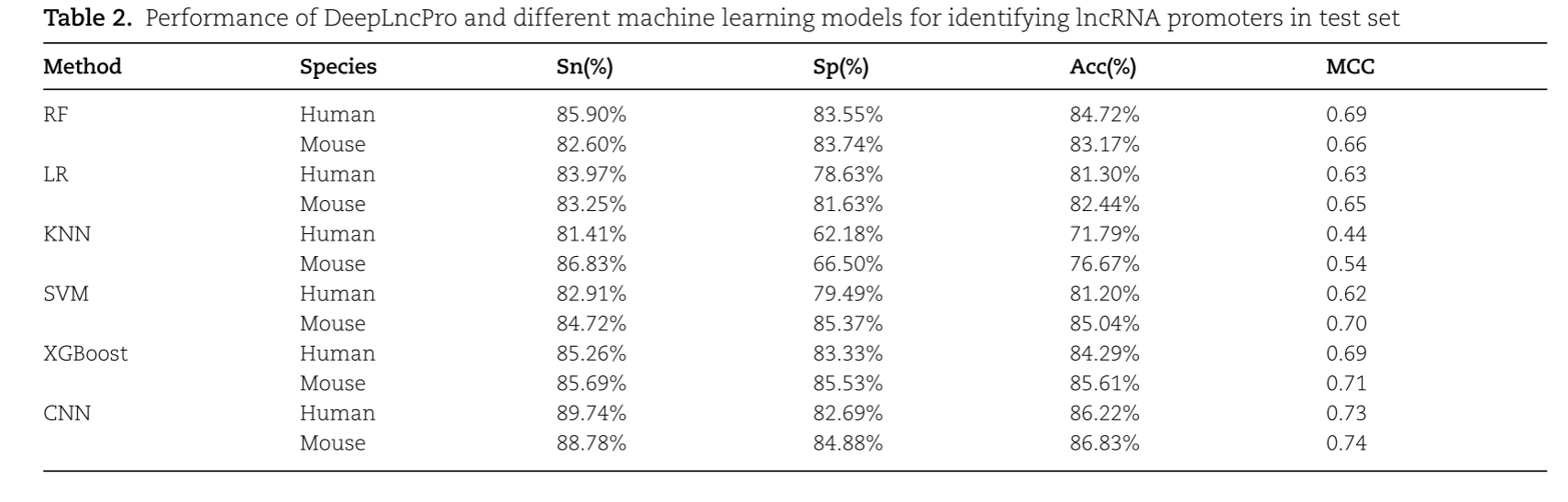
为了证明DeepLncPro在捕获信息基序方面的能力,我们计算了PWM(详见基序提取部分),以分析从第一卷积层的24个过滤器中提取的基序。然后使用TOMTOM将从每个过滤器学习的基序映射到JASPAR数据库中已知的转录因子(TF)结合基序。
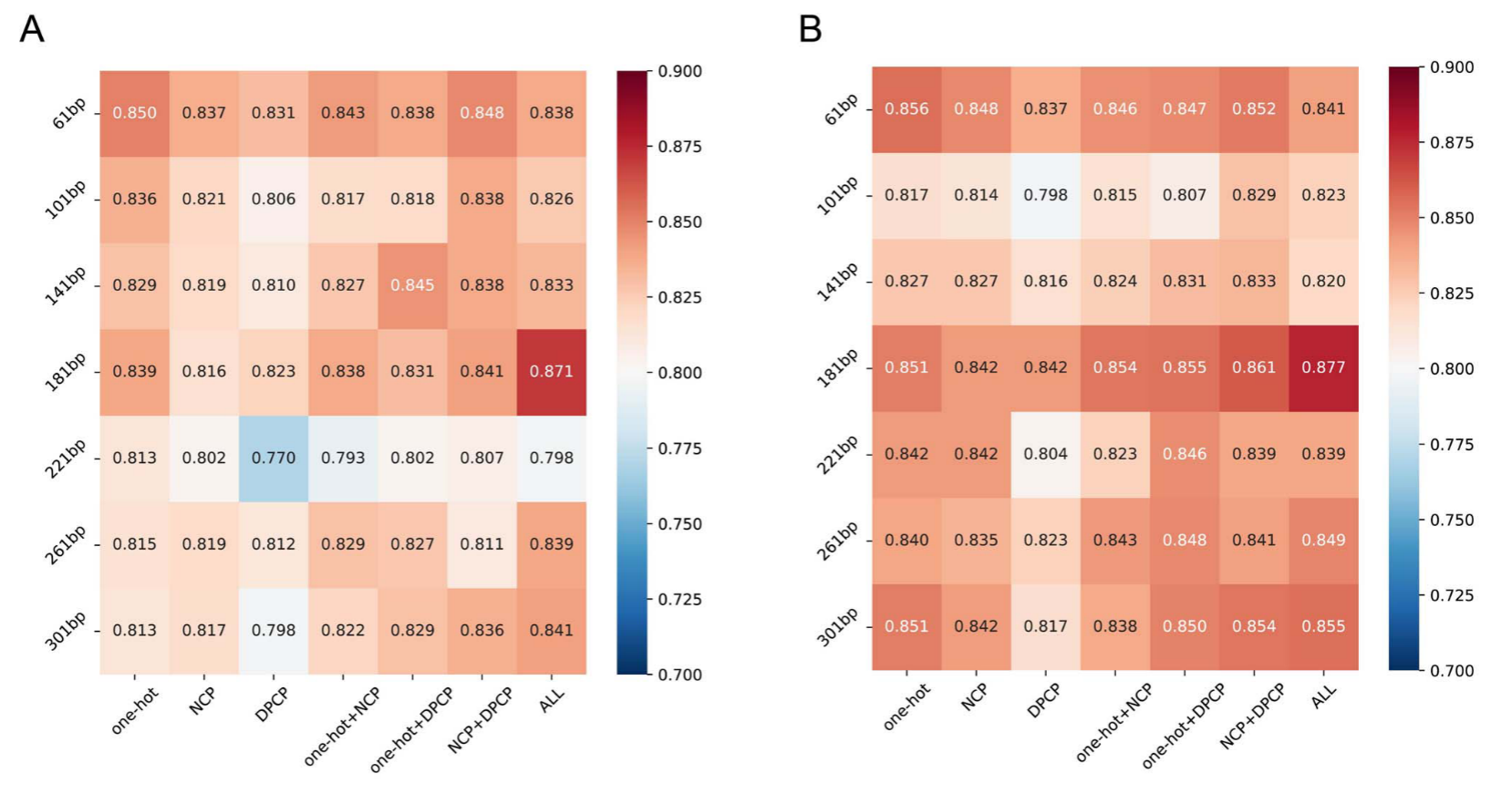
结果表明,基于181 bp序列长度和三种编码方案组合的模型对人和小鼠lncRNA启动子的识别准确率分别为87.07%和87.73%。
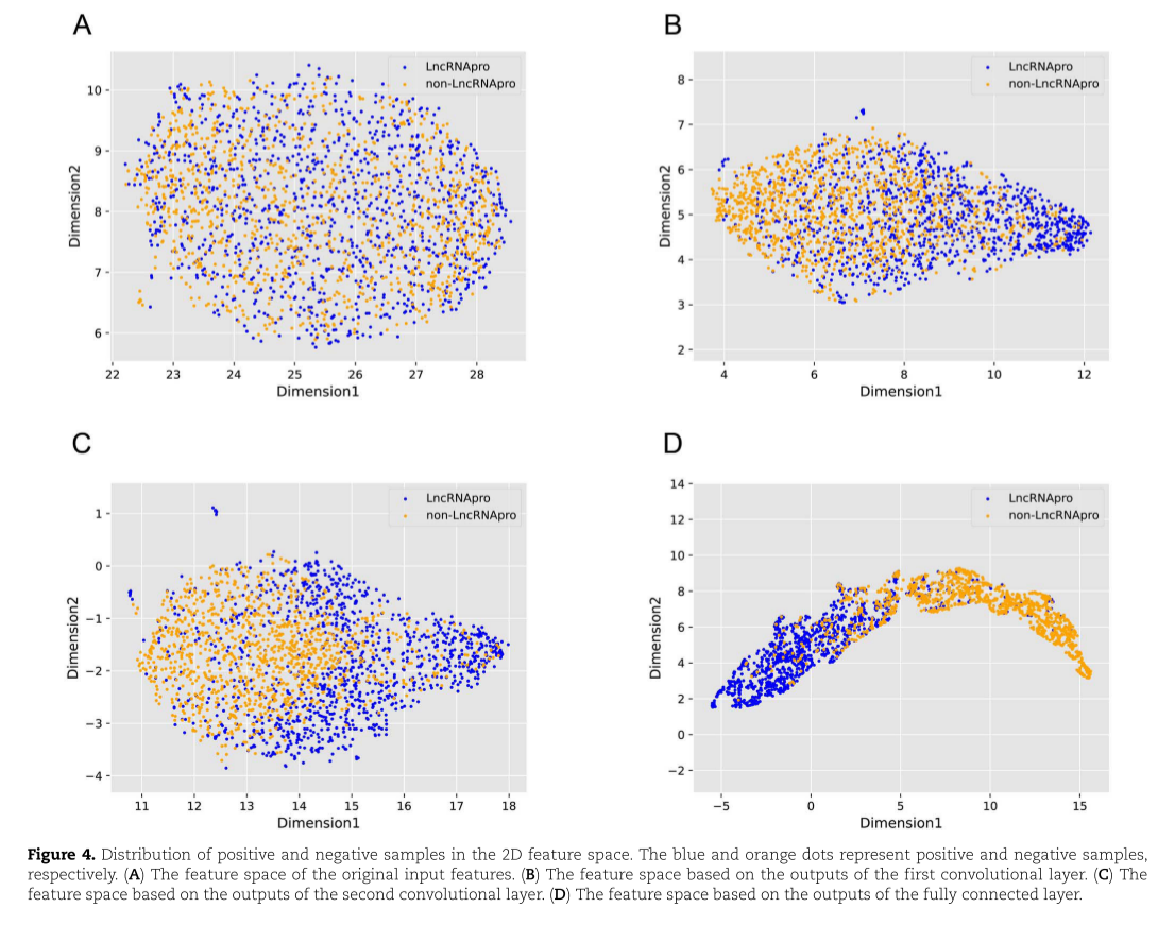
我们提取并可视化了DeepLncPro所有层的输入和输出,即原始输入,第一卷积层的输出,第二卷积层的输出和全连接层的输出。为了便于理解这些特征,使用UMAP [49]显示阳性和阴性样本的分布。
原作者没有给出完整代码,以下是部分代码:
import argparse
import warnings
import torch
import numpy as np
import sys
import torch.nn as nn
import torch.nn.functional as F
import datetime
warnings.filterwarnings("ignore")
def predict(input, model, device):
'''
Overloading DeepLncPro for inference
:return: Model inference results.
'''
model.to(device)
with torch.no_grad():
input = input.to(device)
output = model(input)
pred = output.detach().cpu().numpy().reshape(output.shape[0])
return pred
def motif_check(full_sequence_name, full_sequence, matrix_input_fullseq, model, device, species):
'''
Predicted transcription factor binding sites in sequences.
:param full_sequence_name:List of names of samples.
:param full_sequence:Sequence for each sample.
:param matrix_input_fullseq:A list of input matrices for each sample subsequence.
:return:Results of writing to the output file.
'''
key_filter_dic = {
'm': [0, 4, 7, 9, 12, 16, 17, 18, 20, 23],
'h': [0, 4, 6, 7, 9, 12, 14, 16, 17, 18, 20]
}
bounds_dic = {
'm': [0.7895888715982438, 0.7881207838654518, 0.8854293584823608, 1.3167156040668488, 1.0154612064361572,
1.0301212042570116, 0.9173028439283372, 0.9543586015701295, 0.7805692762136459, 1.1233348727226258],
'h': [0.7927006408572197, 0.7680746629834175, -0.31310261189937594, 0.8761804014444351, 1.316281932592392,
1.0314663887023925, 1.0088603087700905, 1.0300287902355194, 0.922800037264824, 0.9950408935546875,
0.7875141471624375]
}
filter_matched_motif_dic = {
'm': ['MA0746.1', 'MA0810.1', 'MA0079.3', 'MA0140.2', 'MA1125.1', 'MA1106.1', 'MA0140.2', 'MA1125.1',
'MA0746.1', 'MA0140.2'],
'h': ['MA0746.1', 'MA0810.1', 'MA1149.1', 'MA0079.3', 'MA0140.2', 'MA1125.1', 'MA0108.2', 'MA1106.1',
'MA0140.2', 'MA1125.1', 'MA0746.1']
}
motif_TF_dic = {
'm': ['SP3', 'TFAP2A(var.2)', 'SP1', 'GATA1::TAL1', 'ZNF384', 'HIF1A', 'GATA1::TAL1', 'ZNF384', 'SP3',
'GATA1::TAL1'],
'h': ['SP3', 'TFAP2A(var.2)', 'RARA::RXRG', 'SP1', 'GATA1::TAL1', 'ZNF384', 'TBP', 'HIF1A', 'GATA1::TAL1',
'ZNF384', 'SP3']
}
key_filter = key_filter_dic[species]
filter_matched_motif = filter_matched_motif_dic[species]
motif_TF = motif_TF_dic[species]
bounds = bounds_dic[species]
name_list, location_list, sequence_list, motif_list, TF_list = [], [], [], [], []
model.to(device)
for i in range(len(full_sequence_name)):
if len(matrix_input_fullseq[i]) == 0:
continue
input = np.asarray([one_matrix for one_matrix in matrix_input_fullseq[i]], dtype=np.float32)
input = torch.from_numpy(input)
with torch.no_grad():
input = input.to(device)
conv1_out = model.conv1(input).detach().cpu().numpy()
for j in range(len(key_filter)):
num_filter = key_filter[j]
filter_out = conv1_out[:, num_filter, :]
for k in range(len(filter_out[0, :]) - 1):
if filter_out[0, k] > bounds[j]:
name_list.append(full_sequence_name[i])
location_list.append(str(k + 1) + ' to ' + str(k + 10))
sequence_list.append(full_sequence[i][k:k + 10])
motif_list.append(filter_matched_motif[j])
TF_list.append(motif_TF[j])
for l in range(len(filter_out[:, 171])):
if filter_out[l, 171] > bounds[j]:
name_list.append(full_sequence_name[i])
location_list.append(str(l + 172) + ' to ' + str(l + 181))
sequence_list.append(full_sequence[i][l + 171:l + 181])
motif_list.append(filter_matched_motif[j])
TF_list.append(motif_TF[j])
return [name_list, location_list, sequence_list, motif_list, TF_list]
def load_dataset(data_path):
'''
Load the sample sequence from the input file. Perform the process of encoding, sampling, etc.
:param data_path:Input file.
:return:Information used to predict and write to the file.
Include: sample name, sequence, location information, and matrix.
'''
One_hot = {'A': [1, 0, 0, 0],
'T': [0, 1, 0, 0],
'G': [0, 0, 1, 0],
'C': [0, 0, 0, 1]}
NCP = {'A': [1, 1, 1],
'T': [0, 1, 0],
'G': [1, 0, 0],
'C': [0, 0, 1]}
DPCP = {'AA': [0.5773884923447732, 0.6531915653378907, 0.6124592000985356, 0.8402684612384332, 0.5856582729115565,
0.5476708282666789],
'AT': [0.7512077598863804, 0.6036675879079278, 0.6737051546096536, 0.39069870063063133, 1.0,
0.76847598772376],
'AG': [0.7015450873735896, 0.6284296628760702, 0.5818362228429766, 0.6836002897416182, 0.5249586459219764,
0.45903777008667923],
'AC': [0.8257018549087278, 0.6531915653378907, 0.7043281318652126, 0.5882368974116978, 0.7888705476333944,
0.7467063799220581],
'TA': [0.3539063797840531, 0.15795248106354978, 0.48996729107629966, 0.1795369895818257, 0.3059118434042811,
0.32686549630327577],
'TT': [0.5773884923447732, 0.6531915653378907, 0.0, 0.8402684612384332, 0.5856582729115565,
0.5476708282666789],
'TG': [0.32907512978081865, 0.3312861433089369, 0.5205902683318586, 0.4179453841534657, 0.45898067049412195,
0.3501900760908136],
'TC': [0.5525570698352168, 0.6531915653378907, 0.6124592000985356, 0.5882368974116978, 0.49856742124957026,
0.6891727614587756],
'GA': [0.5525570698352168, 0.6531915653378907, 0.6124592000985356, 0.5882368974116978, 0.49856742124957026,
0.6891727614587756],
'GT': [0.8257018549087278, 0.6531915653378907, 0.7043281318652126, 0.5882368974116978, 0.7888705476333944,
0.7467063799220581],
'GG': [0.5773884923447732, 0.7522393476914946, 0.5818362228429766, 0.6631651908463315, 0.4246720956706261,
0.6083143907016332],
'GC': [0.5525570698352168, 0.6036675879079278, 0.7961968911255676, 0.5064970193495165, 0.6780274730118172,
0.8400043540595654],
'CA': [0.32907512978081865, 0.3312861433089369, 0.5205902683318586, 0.4179453841534657, 0.45898067049412195,
0.3501900760908136],
'CT': [0.7015450873735896, 0.6284296628760702, 0.5818362228429766, 0.6836002897416182, 0.5249586459219764,
0.45903777008667923],
'CG': [0.2794124572680277, 0.3560480457707574, 0.48996729107629966, 0.4247569687810134, 0.5170412957708868,
0.32686549630327577],
'CC': [0.5773884923447732, 0.7522393476914946, 0.5818362228429766, 0.6631651908463315, 0.4246720956706261,
0.6083143907016332]}
name_list, sequence_list, matrix_list = [], [], []
with open(data_path, 'rt') as f:
lines = f.readlines()
if lines[0][0] != '>':
print('Please check the input file format!')
sys.exit()
k = 0
sequence = ''
for line in lines:
if line[0] == '>':
name_list.append(line[1:].strip())
if k == 1:
sequence_list.append(sequence)
sequence = ''
k = 1
else:
sequence = sequence + line.strip()
sequence_list.append(sequence)
name_output, location_output, sequence_output = [], [], []
matrix_oneseq_all = []
#Sampling with sliding windows.
for i in range(len(name_list)):
matrix_oneseq = []
name_cur = name_list[i]
sequence_cur = sequence_list[i]
if len(sequence_cur) < 181:
print('The input sequence ''%s'' does not meet the minimum length requirement of 181bp.' % name_list[i])
else:
# Slide the middle part of the window.
matrix_middle = np.zeros([13, 178])
for pos in range(178):
matrix_middle
matrix_middle[0:4, pos] += np.asarray(np.float32(One_hot[sequence_cur[pos + 1]]))
matrix_middle[4:7, pos] += np.asarray(np.float32(NCP[sequence_cur[pos + 1]]))
matrix_middle[7:13, pos] += np.asarray(np.float32(DPCP[sequence_cur[pos:pos + 2]])) / 2
matrix_middle[7:13, pos] += np.asarray(np.float32(DPCP[sequence_cur[pos + 1:pos + 3]])) / 2
#The changes of sliding window.
for left in range(len(sequence_cur) - 180):
right = left + 180
name_output.append(name_cur)
location_output.append(str(left + 1) + ' to ' + str(left + 181))
sequence_output.append(sequence_cur[left:right + 1])
matrix_left = np.concatenate((np.asarray(
np.float32(One_hot[sequence_cur[left]] + NCP[sequence_cur[left]])),
np.asarray(np.float32(DPCP[sequence_cur[left:left + 2]])) / 2),
axis=0)
matrix_right1 = np.concatenate(
(np.asarray(np.float32(One_hot[sequence_cur[right - 1]] + NCP[sequence_cur[right - 1]])),
np.asarray(np.float32(DPCP[sequence_cur[right - 2:right]])) / 2 +
np.asarray(np.float32(DPCP[sequence_cur[right - 1:right + 1]])) / 2),
axis=0)
matrix_right2 = np.concatenate((np.asarray(
np.float32(One_hot[sequence_cur[right]] + NCP[sequence_cur[right]])), np.asarray(
np.float32(DPCP[sequence_cur[right - 1:right + 1]])) / 2), axis=0)
matrix_right = np.concatenate((matrix_right1[:, np.newaxis], matrix_right2[:, np.newaxis]), axis=1)
matrix_cur = np.concatenate((matrix_left[:, np.newaxis], matrix_middle, matrix_right), axis=1)
matrix_middle = matrix_cur[:, 2:180]
matrix_list.append(matrix_cur)
matrix_oneseq.append(matrix_cur)
matrix_oneseq_all.append(matrix_oneseq)
matrix_input = np.asarray([i for i in matrix_list], dtype=np.float32)
matrix_input = torch.from_numpy(matrix_input)
return name_list, sequence_list, matrix_oneseq_all, name_output, location_output, sequence_output, matrix_input
class DeepncPro(nn.Module):
def __init__(self, configs):
super(DeepncPro, self).__init__()
self.sequence_lenth = configs.sequence_lenth
self.input_channel = configs.input_channel
self.out_channel1 = configs.out_channel1
self.out_channel2 = configs.out_channel2
self.filter_size = configs.filter_size
self.filter_size2 = configs.filter_size2
self.stride = configs.stride
self.fc1_size = configs.fc1_size
self.fc2_size = configs.fc2_size
self.pool_1 = configs.pool_1
self.pool_2 = configs.pool_2
self.fc2 = configs.fc2
flatten_size = (self.sequence_lenth - self.filter_size) // self.stride + 1
if configs.pool_1:
flatten_size = (flatten_size - self.filter_size2) // self.stride + 1
flatten_size = (flatten_size - self.filter_size2) // self.stride + 1
if configs.pool_2:
flatten_size = (flatten_size - self.filter_size2) // self.stride + 1
self.flatten_size = flatten_size
self.conv1 = nn.Conv1d(in_channels=self.input_channel, out_channels=self.out_channel1,
kernel_size=self.filter_size, stride=self.stride)
self.max_pool1 = nn.MaxPool1d(kernel_size=self.filter_size2, stride=self.stride)
self.conv2 = nn.Conv1d(self.out_channel1, self.out_channel2, self.filter_size2, self.stride)
self.max_pool2 = nn.MaxPool1d(self.filter_size2, self.stride)
self.liner1 = nn.Linear(self.out_channel2 * self.flatten_size, self.fc1_size)
self.liner2 = nn.Linear(self.fc1_size, self.fc2_size)
if configs.fc2:
self.liner3 = nn.Linear(self.fc2_size, 1)
else:
self.liner3 = nn.Linear(self.fc1_size, 1)
def forward(self, x):
x = F.relu(self.conv1(x))
if self.pool_1:
x = self.max_pool1(x)
x = F.relu(self.conv2(x))
if self.pool_2:
x = self.max_pool2(x)
x = x.view(x.size(0), -1)
x = F.relu(self.liner1(x))
if self.fc2:
x = F.relu(self.liner2(x))
x = self.liner3(x)
return torch.sigmoid(x)
def prediction_process(input_file, species):
'''
Prediction of input sequences using DeepLncPro model.
:return: List of predicted results.
'''
device = torch.device("cuda:0" if torch.cuda.is_available() else "cpu")
model = torch.load('model/DeepLncPro.pkl', map_location='cpu')
full_sequence_name, full_sequence, matrix_input_fullseq, name_list, location_list, sequence_list, data_list = load_dataset(
input_file)
pre_list = predict(data_list, model, device)
out_list1 = [name_list, location_list, sequence_list, pre_list]
out_list2 = motif_check(full_sequence_name, full_sequence, matrix_input_fullseq, model, device, species)
return out_list1, out_list2
def write_outputFile(output_list, outputFile, threshold):
'''Write the results to outputFile.'''
f = open(outputFile, 'w', encoding="utf-8")
name_list = output_list[0]
location_list = output_list[1]
sequence_list = output_list[2]
pre_list = output_list[3]
prediction_list = [1 if i > threshold else 0 for i in pre_list]
out1 = open("js/out1.txt", "r")
out2 = open("js/out2.txt", "r")
s = out1.read()
f.write(s)
out1.close()
for i in range(len(name_list)):
f.write('<tr>' + '\n')
f.write('<td>' + str(i + 1) + '</td>' + '\n')
f.write('<td>' + name_list[i] + '</td>' + '\n')
f.write('<td>' + location_list[i] + '</td>' + '\n')
f.write('<td>' + str(pre_list[i]) + '</td>' + '\n')
f.write('<td>' + str(prediction_list[i]) + '</td>' + '\n')
f.write('<td>' + sequence_list[i] + '</td>' + '\n')
f.write('</tr>' + '\n')
s = out2.read()
f.write(s)
out2.close()
f.close()
def write_motif_check_results(output_list, outputFile):
'''Write the results to outputFile.'''
f = open(outputFile, 'w', encoding="utf-8")
name_list = output_list[0]
location_list = output_list[1]
sequence_list = output_list[2]
motif_list = output_list[3]
TF_list = output_list[4]
out1 = open("js/out3.txt", "r")
out2 = open("js/out4.txt", "r")
s = out1.read()
f.write(s)
out1.close()
for i in range(len(name_list)):
f.write('<tr>' + '\n')
f.write('<td>' + str(i + 1) + '</td>' + '\n')
f.write('<td>' + name_list[i] + '</td>' + '\n')
f.write('<td>' + location_list[i] + '</td>' + '\n')
f.write('<td>' + sequence_list[i] + '</td>' + '\n')
f.write('<td>' + motif_list[i] + '</td>' + '\n')
f.write('<td>' + TF_list[i] + '</td>' + '\n')
f.write('</tr>' + '\n')
s = out2.read()
f.write(s)
out2.close()
f.close()
def parse_args():
'''Parameters.'''
description = "DeepLncPro is able to identify the promoter of non-coding RNA in Human and Mouse.\n" \
"Example: python DeepLncPro.py -i example.txt -o output.html -s Human -ts 0.5"
parser = argparse.ArgumentParser(description=description)
parser.add_argument('-i', '--inputFile',
help='-i example.txt (The input file is a complete Fasta format sequence.)')
parser.add_argument('-o1', '--outputFile1',
help='-o1 output_prediction.html (Results of predicting lncRNA promoters are saved under results folder.)')
parser.add_argument('-o2', '--outputFile2',
help='-o2 output_motifcheck.html (Results of predicting motifs are saved under results folder.')
parser.add_argument('-s', '--species',
help='-s h/m (Choose Human/Mouse from two species to use.)')
parser.add_argument('-ts', '--threshold',
help='-ts 0.5(Prediction result threshold)')
args = parser.parse_args()
return args
def preprocess(inputFile, outputFile1, outputFile2, species, threshold):
'''Predicting lncRNA promoters and writing output files.'''
print(datetime.datetime.now())
out_list1, out_list2 = prediction_process(inputFile, species)
write_outputFile(out_list1, outputFile1, threshold)
write_motif_check_results(out_list2, outputFile2)
print(datetime.datetime.now())
if __name__ == '__main__':
args = parse_args()
preprocess(args.inputFile, args.outputFile1, args.outputFile2, args.species, float(args.threshold))


.png)